ADMET Properties¶
Every PocketDock job ships with an ADMET / drug-likeness panel on the results page. Properties are computed automatically from the uploaded ligand with RDKit — no opt-in, no extra cost in runtime. The panel sits alongside the docking poses so you can sanity-check a candidate's drug-likeness at the same time you're inspecting its binding pose.
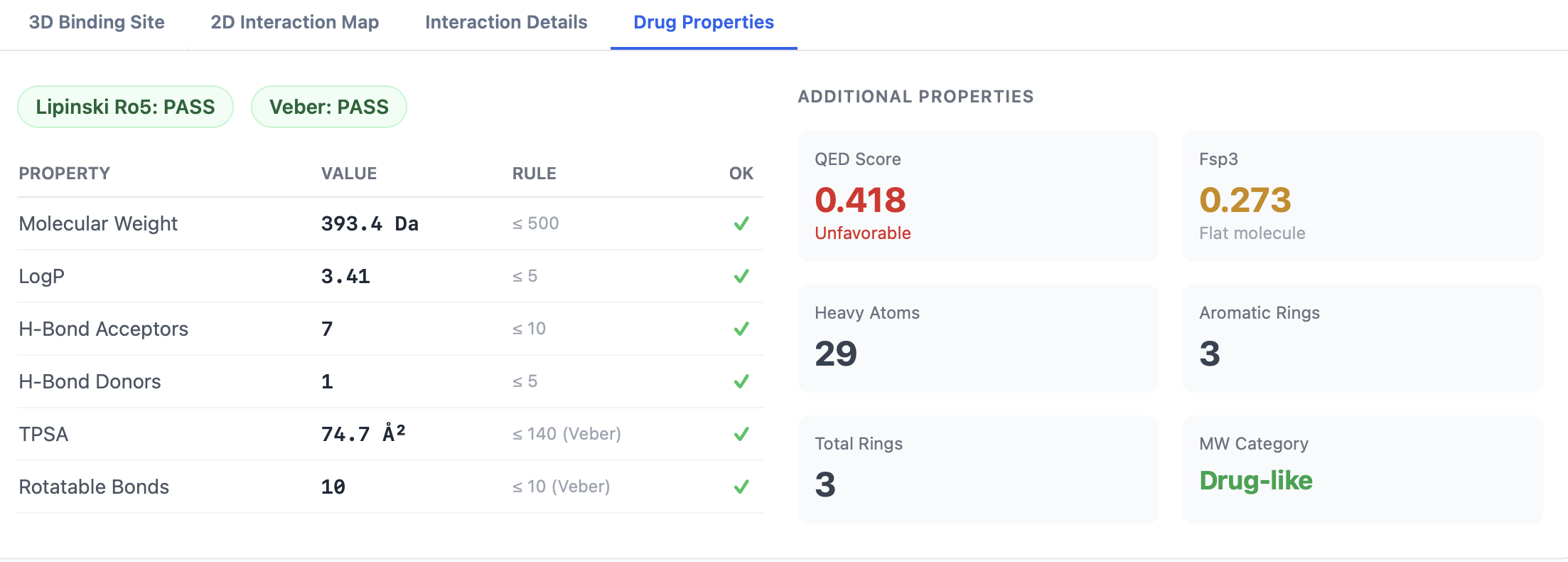
What gets computed¶
PocketDock computes a focused set of RDKit descriptors that map onto common medicinal-chemistry filters:
| Descriptor | RDKit source | Typical range / notes |
|---|---|---|
| MW | Molecular weight (Da) | Lipinski threshold: ≤ 500 |
| logP | Crippen logP (octanol/water partition) | Lipinski threshold: ≤ 5 |
| HBA | H-bond acceptor count | Lipinski threshold: ≤ 10 |
| HBD | H-bond donor count | Lipinski threshold: ≤ 5 |
| TPSA | Topological polar surface area (Ų) | Veber threshold: ≤ 140 |
| Rotatable bonds | Number of rotatable bonds | Veber threshold: ≤ 10 |
| Aromatic rings | Count of aromatic rings | — |
| Heavy atoms | Non-hydrogen atom count | Used in ligand-efficiency calculation |
| Ring count | Total ring count | — |
| Fsp3 | Fraction of sp³ carbons | Higher Fsp3 ≈ more three-dimensional, often better drug-like |
| QED | Quantitative Estimate of Drug-likeness (Bickerton 2012) | [0, 1]. ~0.5–0.9 is typical for marketed drugs. |
| Lipinski violations | Count of broken Lipinski rules (0–4) | 0 is best; ≤ 1 traditionally tolerated |
| Lipinski pass | true if 0 violations |
Boolean badge |
| Veber pass | true if TPSA ≤ 140 and rotatable bonds ≤ 10 |
Boolean badge |
Lipinski's rule of five¶
Christopher Lipinski's classic filter for oral bioavailability says a drug-like molecule should not violate more than one of:
- MW ≤ 500 Da
- logP ≤ 5
- H-bond donors ≤ 5
- H-bond acceptors ≤ 10
PocketDock counts how many of these four rules your ligand breaks. The Lipinski pass badge is green when the count is 0.
Failing Lipinski is not failing biology
Plenty of marketed drugs violate one or even two rules. Use Lipinski as a coarse triage filter for orally administered small molecules, not as an absolute go/no-go.
Veber's rules¶
Veber's extension covers oral bioavailability from a different angle:
- TPSA ≤ 140 Ų
- Rotatable bonds ≤ 10
The Veber pass badge requires both conditions. Veber tends to flag flexible molecules and overly polar ones — useful when Lipinski misses something obvious.
QED — a single drug-likeness number¶
QED (Bickerton et al., 2012) collapses MW, logP, HBA, HBD, PSA, rotatable bonds, aromatic rings, and structural alerts into one number on [0, 1]. Rough mental model:
| QED | Interpretation |
|---|---|
≥ 0.8 |
Highly drug-like |
0.5–0.8 |
Drug-like — most marketed oral drugs sit here |
0.3–0.5 |
Borderline; passes some filters and fails others |
< 0.3 |
Looks unusual for an oral drug — could still be a tool compound or non-oral candidate |
QED is the most useful single number on the panel for ranking analogues against each other.
Supported ligand formats¶
ADMET computation runs the standard RDKit ligand-parsing path:
.sdf(preferred).mol2.mol
If RDKit fails to parse the ligand (bad valences, unusual atoms, missing 3D coordinates), the panel will be empty and the admet field in the API response will be {}. The docking pipeline still runs — ADMET is computed in parallel and failures are non-fatal. Check the worker logs if the panel is unexpectedly blank.
Where to find it¶
On the results page¶
The ADMET panel sits below the pockets summary. Each numeric value is shown alongside the relevant threshold; the Lipinski and Veber badges are colored green/red according to pass/fail.
In the API¶
GET /api/jobs/<job_id>/results/ includes an admet dict with all descriptors:
import requests
results = requests.get("http://localhost:8000/api/jobs/42/results/").json()
admet = results["admet"]
print(f"MW = {admet['mw']:.1f} Da, logP = {admet['logp']:.2f}, QED = {admet['qed']:.2f}")
print(f"Lipinski pass: {admet['lipinski_pass']} Veber pass: {admet['veber_pass']}")
See the API reference for the full response schema.
In batch screening¶
The batch dashboard does not show per-ligand ADMET — open an individual ligand's results page to see its panel, or fetch the results JSON for each job and aggregate client-side. A typical post-screening filter:
hits = []
for job in batch["jobs"]:
if job["status"] != "completed":
continue
r = requests.get(f"http://localhost:8000/api/jobs/{job['id']}/results/").json()
admet = r.get("admet", {})
if admet.get("lipinski_pass") and admet.get("qed", 0) >= 0.5:
hits.append((job["ligand_name"], job["best_score"], admet["qed"]))
hits.sort(key=lambda x: (-x[1], -x[2]))
Tips¶
- Sort batch hits by QED × combined score when you want a single ranking that balances binding and drug-likeness.
- Don't reject borderline Lipinski violations outright — peptidomimetics, macrocycles, and PROTACs deliberately leave Lipinski space.
- Watch for Fsp3 — a low Fsp3 (highly flat, aromatic molecule) often correlates with poor solubility and high promiscuity, even if everything else looks fine.
- TPSA matters for CNS targets — for blood-brain-barrier penetration the practical threshold is closer to TPSA ≤ 90 Ų, well below Veber's 140.
- The panel is descriptors, not ADME predictions — no permeability, clearance, or toxicity model. For those, plug the ligand into a dedicated ADMET predictor.
See also¶
- Concepts — terminology refresher
- Interpreting Results — ADMET — how to read each row of the panel in context
- Batch Docking — combining docking ranks with ADMET filters in screening