Batch Docking¶
Batch docking lets you screen many ligands against the same protein in a single submission. PocketDock creates one DockingJob per ligand, ties them together with a shared batch ID, and shows progress and per-ligand best scores on a dedicated dashboard.
When to use it¶
- Library screening — dock 10–100 candidates against the same target and rank by combined score.
- Series exploration — submit an SD file from your chemistry team and let PocketDock split it into one job per molecule automatically.
- Reproducibility — every ligand in the batch uses the same protein, pockets, exhaustiveness, and feature flags, so comparisons are apples-to-apples.
If you only have one or two ligands, the single-job tab is simpler — there's no batch overhead and the results page is the same.
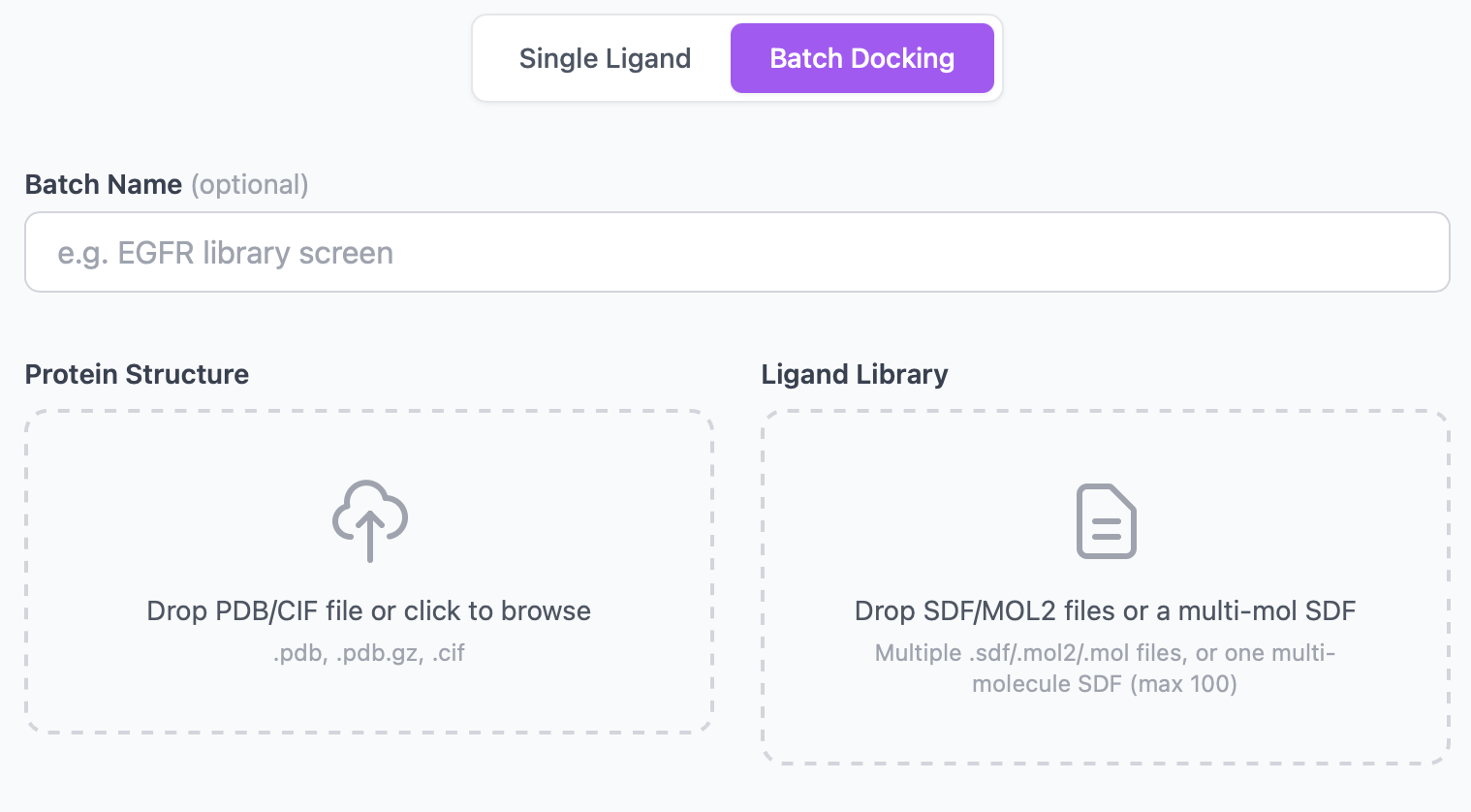
Submitting a batch¶
The upload page has two tabs: Single Job and Batch. Switch to Batch and you'll get:
| Field | Required | Notes |
|---|---|---|
| Batch name | No | Free-text label. Shows in the dashboard header; defaults to empty. |
| Protein file | Yes | Same formats as single-job (.pdb, .pdb.gz, .cif, ≤ 50 MB). |
| Ligand files | Yes | One or more .sdf, .mol2, or .mol files. Up to 100 files per batch. Each ≤ 10 MB. |
| Number of pockets | No | 1–20, default 3. Applied to every ligand. |
| Vina exhaustiveness | No | 1–64, default 8. Applied to every ligand. |
| Scoring function | No | vina (default) or vinardo. |
| Refine poses | No | If checked, every job runs OpenMM minimization on its poses. |
| MM-GBSA rescore | No | If checked, every job adds an MM-GBSA-style ΔG column. |
| Ensemble | No | Optional — combines with batch (every ligand is docked across the same ensemble). |
Multi-molecule SDF files¶
If a ligand file is an SDF containing more than one molecule (separated by $$$$), PocketDock splits it automatically and creates one job per molecule. The molecule's title line (first line of the SDF block) is used as the ligand name; falling back to mol_1, mol_2, … if the title is blank.
You can mix and match: drop a 50-molecule library SDF and three single-molecule SDFs in the same batch — you'll get 53 jobs.
File limits at a glance¶
| Limit | Value |
|---|---|
| Maximum ligand files per submission | 100 |
| Maximum per-file size | 10 MB |
| Maximum protein size | 50 MB |
If you exceed any limit, the form returns a validation error and nothing is submitted.
The batch dashboard¶
After submission you're redirected to /batch/<batch_id>/. The dashboard shows:
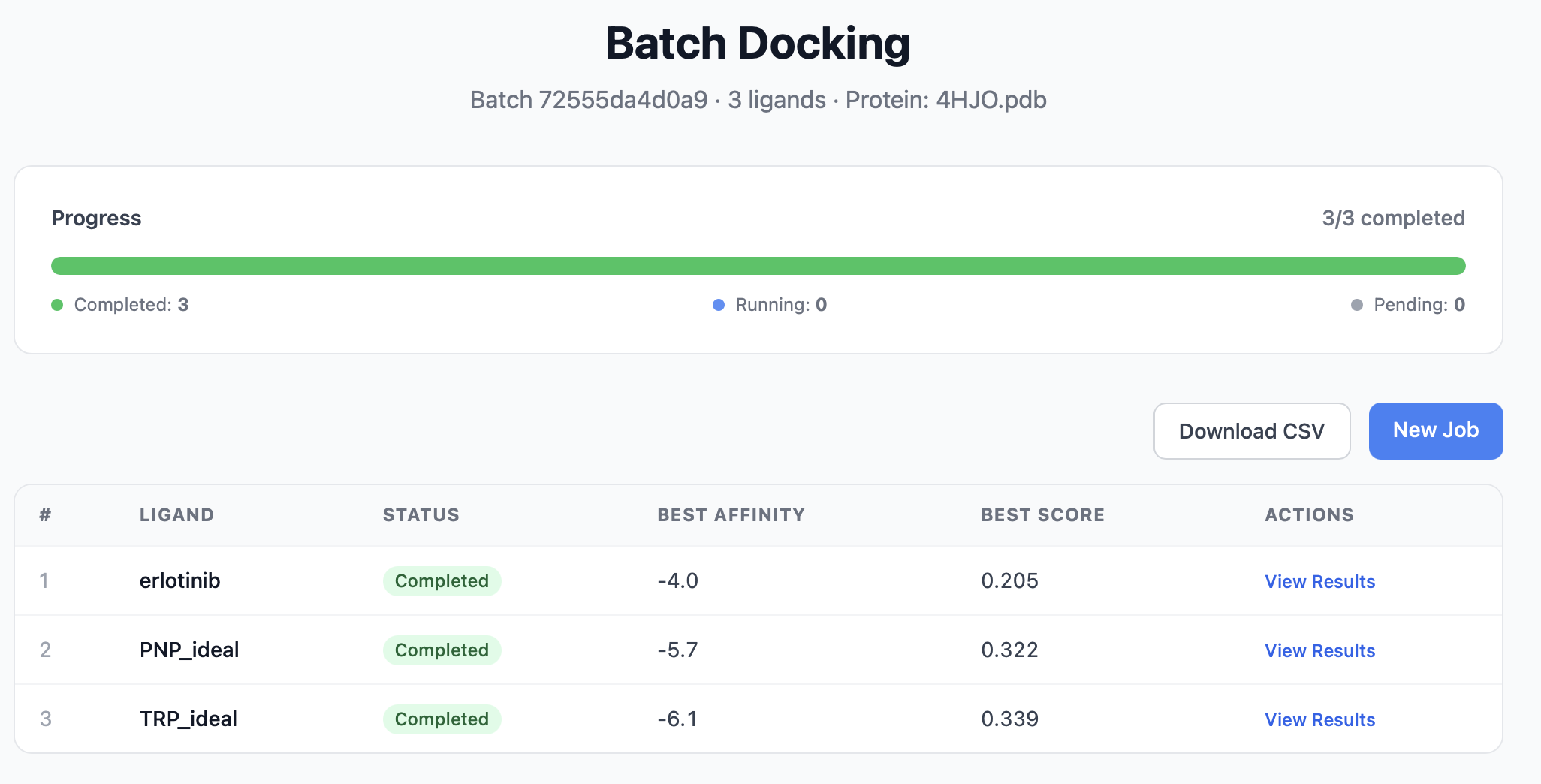
| Panel | Contents |
|---|---|
| Header | Batch name, protein filename, batch ID (the URL slug) |
| Progress bar | Total / completed / failed / running / pending counts and a percentage |
| Per-ligand table | One row per ligand: status, best Vina affinity, best combined score, link to that ligand's full results page |
The page auto-refreshes via the /api/batch/<batch_id>/ endpoint, so you can leave it open and watch the batch finish. When all_done is true, the progress bar locks in.
Each row links to that ligand's regular results page — the per-ligand view is exactly the same as a single-job result, with the ADMET panel, 3D viewer, and interaction analysis.
Ranking the hits¶
Sort the dashboard table by best combined score to see which ligands docked best overall, or by best affinity to rank by Vina score alone. For programmatic ranking, fetch /api/batch/<batch_id>/ and sort the jobs array — see the API reference.
A typical workflow:
- Sort by best combined score, descending.
- Inspect the top 5–10 ligands by opening each results page.
- For survivors, check specific interactions (H-bonds, salt bridges with conserved residues) — a high score with no specific interactions is a red flag (see Interpreting Results).
- For survivors that passed step 3, check the ADMET panel for any non-starter properties (Lipinski violations, low QED).
How it works under the hood¶
- Submitting the batch form creates one
DockingJobper ligand, all sharing the samebatch_id(a 12-char UUID slice). - Each job has its own
job_diron disk, ligand name, and Celery task. They run independently and may complete out of order. - The dashboard page queries
DockingJob.objects.filter(batch_id=batch_id)and aggregates the rows. - If you want to fan out faster, scale the Celery worker (more replicas or higher
--concurrency— see Configuration).
Tips¶
- Pre-clean your library — invalid SDFs (bad valences, missing atoms) fail at the RDKit / Meeko parse step and show up as
failedrows on the dashboard with a reason in the per-job error message. - Name your molecules — set the SDF title line; it becomes the dashboard label. Anonymous molecules end up as
mol_1,mol_2, … and are hard to track. - Start small — run a 5-ligand pilot before submitting 100. It catches preparation issues fast and gives you a runtime estimate.
- Disable expensive options for screening — leave refine poses and MM-GBSA rescore off for first-pass screening (they multiply runtime per job); turn them on for the shortlist.