Interaction Analysis¶
Each docked pose comes with an automatic geometric analysis of the protein–ligand contacts. PocketDock detects six interaction types, visualizes them in 3D, summarizes them in a 2D map, and lists every contact in a sortable table.
The six interaction types¶
PocketDock detects interactions by geometry — distance and angle thresholds between specific atom types. Defaults are chosen to match the conservative end of the published literature.
| Type | Default cutoff | Color in viewer | Notes |
|---|---|---|---|
| H-bond | 3.5 Å | Yellow | Donor–acceptor distance; angle filtering applied |
| Hydrophobic | 4.0 Å | Green | Carbon–carbon contacts in non-polar environments |
| Salt bridge | 4.0 Å | Pink | Charged-group pairs (cationic + anionic) |
| π-stacking | 5.5 Å | Cyan | Aromatic ring centroid–centroid distance |
| π-cation | 6.0 Å | Purple | Aromatic ring + adjacent cationic group |
| Halogen bond | 4.0 Å | Red | Halogen donor → O/N acceptor |
In the 3D viewer, each detected interaction is a dashed line of the corresponding color, with a distance label.

Customizing thresholds¶
Each interaction type has its own slider in the Interaction controls panel. Drag a slider to change the cutoff and the viewer updates immediately — interactions outside the new threshold disappear.
A Reset to defaults button restores all thresholds in one click.
Near-miss toggle¶
Sometimes a contact is just outside the cutoff and you want to see it anyway. Enable Near-miss to also draw interactions within 120% of each threshold (e.g., H-bonds out to 4.2 Å when the cutoff is 3.5 Å). Near-miss interactions are drawn with a dashed-and-faded line so you can tell them apart from true contacts.
The 2D Interaction Map tab¶
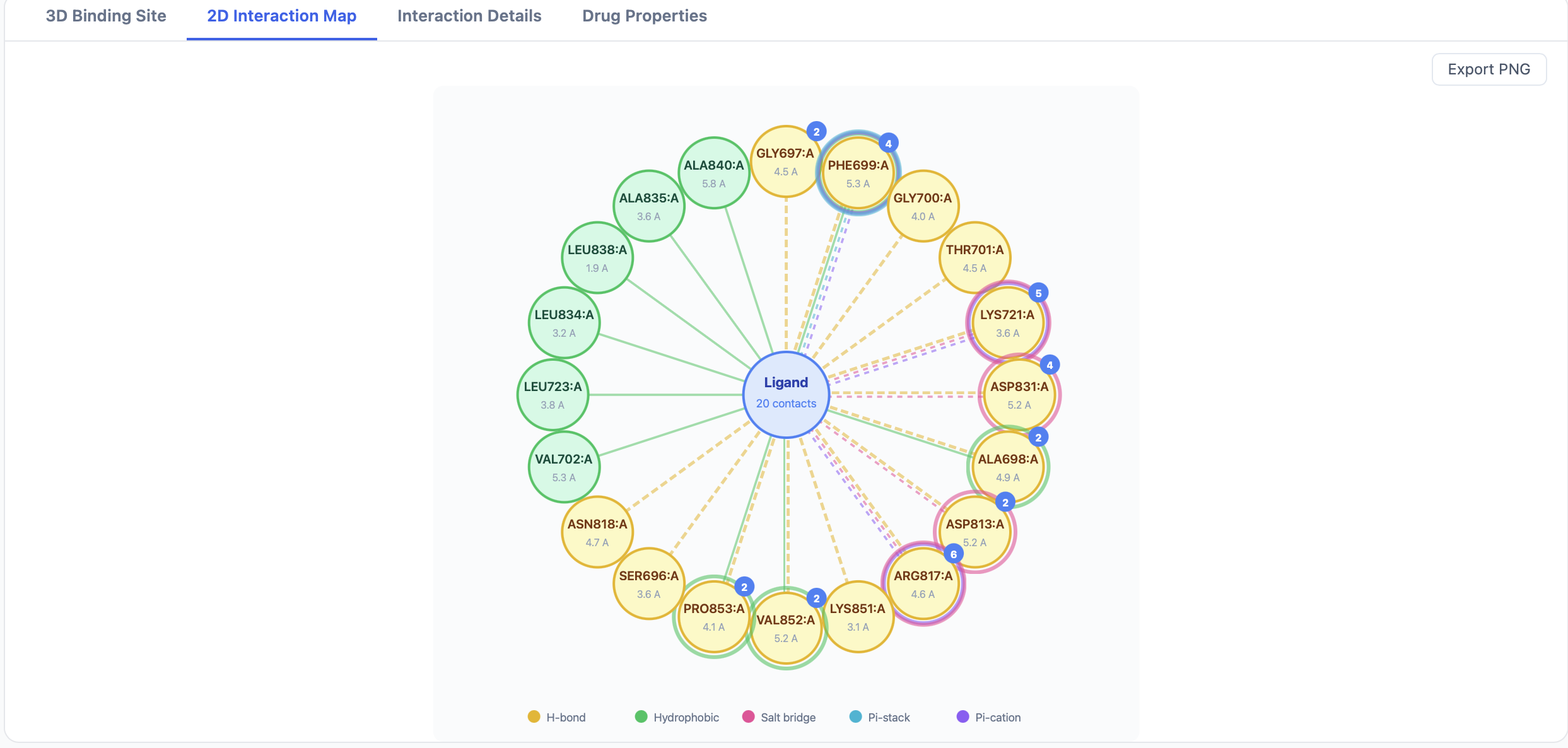
The 2D map renders the ligand at the center with the contacting residues arranged around it, LigPlot-style. Each interaction line is colored using the same scheme as the 3D viewer:
- Distance is annotated on the line.
- Residue labels include the three-letter code, residue number, and chain.
- Aromatic π-interactions are drawn as ring–ring or ring–cation linkages.
Use Export 2D Map PNG at the top of the results page to save the current map as an image.
The Interaction Details tab¶
The Details tab is a sortable, filterable table — one row per detected interaction:
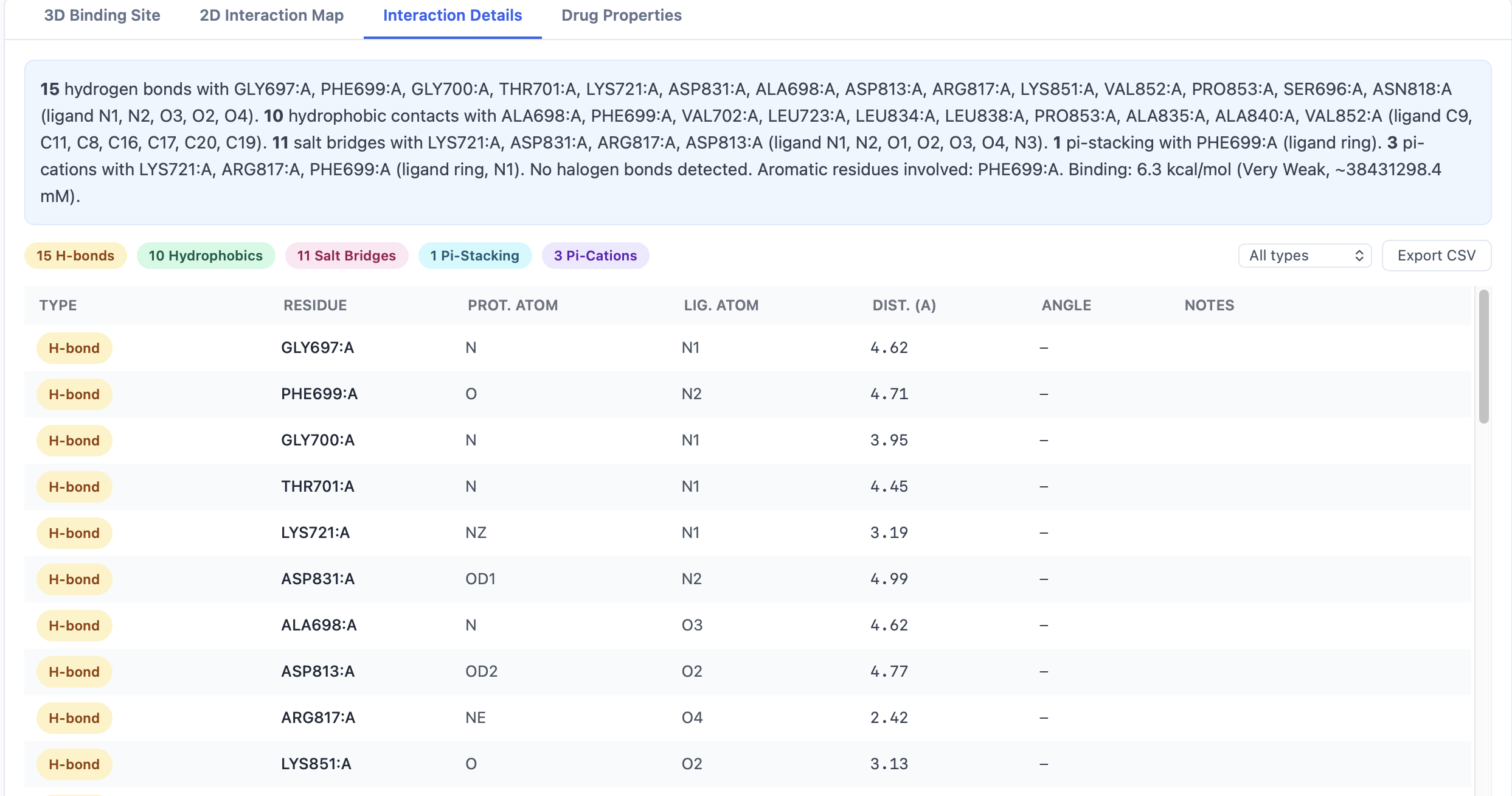
| Column | Content |
|---|---|
| Type | H-bond, hydrophobic, salt bridge, π-stack, π-cation, halogen |
| Ligand atom | Atom name (e.g., O3) |
| Residue | Three-letter residue code + number + chain (e.g., ASP142.A) |
| Distance (Å) | Atom-to-atom or centroid-to-centroid distance |
A row of filter buttons above the table narrows the view:
- All — every interaction
- Aromatic only — π-stacking and π-cation
- H-bonds only — H-bonds and halogen bonds
- Charged only — salt bridges and π-cation
The Export Interactions CSV button exports the currently filtered view.
When interaction detection fails silently¶
The interaction detector runs after Vina finishes and is wrapped in error handling — if it crashes on a particular pose (for example, an unusual residue type that can't be parsed), the failure is logged but does not kill the job. The pose still appears in the results table; it just won't have interactions associated with it. Check the worker logs if you see a pose with no interaction lines and you expected some.
Tips¶
- Start with defaults. PocketDock's cutoffs are calibrated for typical drug-like ligands. Loosening them helps you spot weak contacts; tightening them produces tighter interaction sets for figures.
- Use near-miss to find borderline H-bonds. Crystallographic uncertainty alone is often ±0.3 Å; a putative H-bond at 3.7 Å is plausible but missed by the default 3.5 Å cutoff.
- Filter to "Aromatic only" when you're investigating π-stacking with kinase hinge residues or analogous aromatic clusters.
- Compare poses by sorting interactions by type and counting — a pose with 2 H-bonds and 5 hydrophobic contacts may be more credible than one with 0 H-bonds and 8 hydrophobic, even at the same affinity.