Uploading a Job¶
The upload page is the entry point to PocketDock. It's a single form with two file drop zones and an optional advanced settings panel.
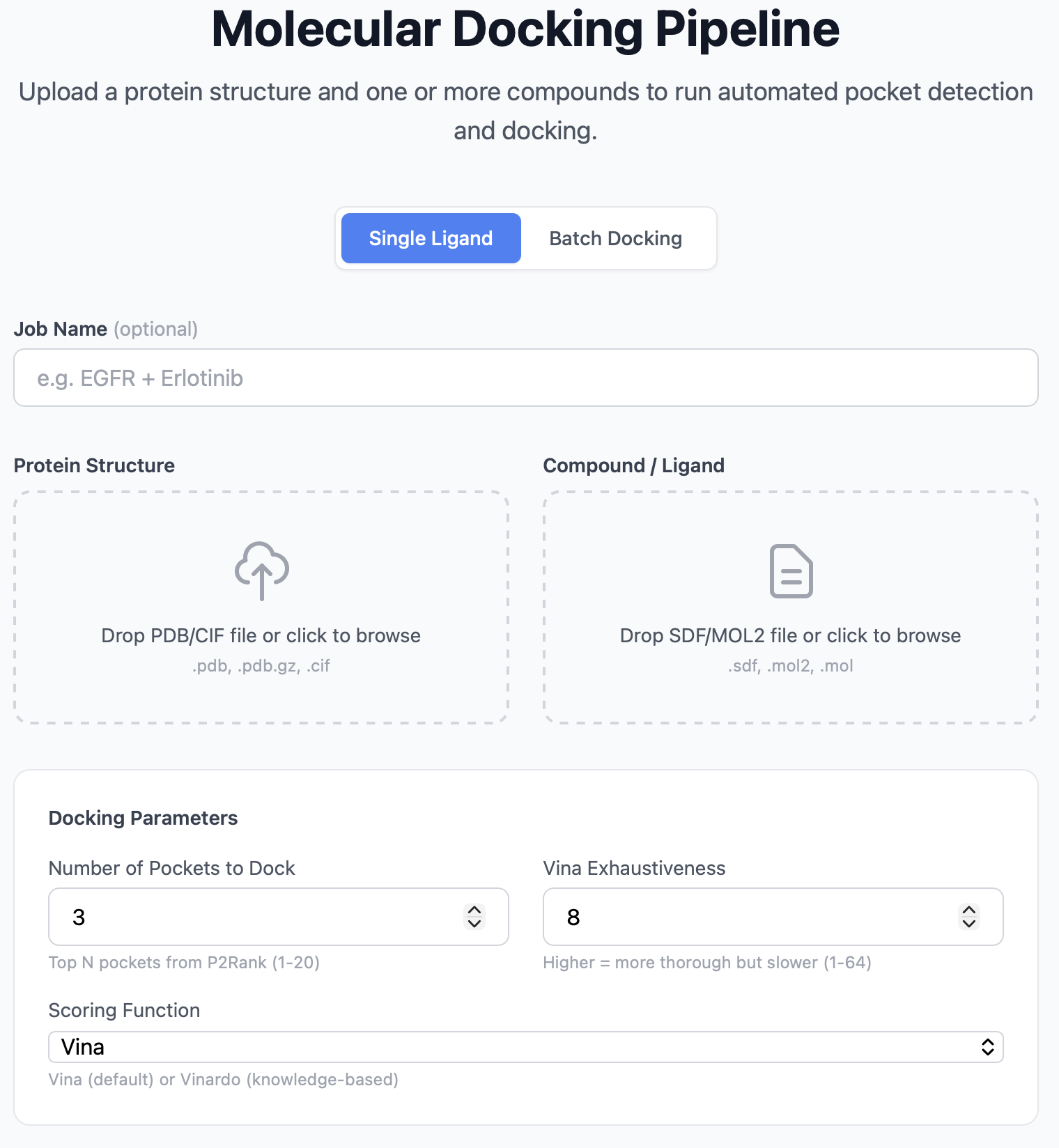
Form fields¶
| Field | Required | Notes |
|---|---|---|
| Job name | No | Free-text label for your records. Defaults to <protein>_<ligand> if blank. |
| Protein file | Yes | The receptor structure. See supported formats below. |
| Ligand file | Yes | The small molecule to dock. See supported formats below. |
| Number of pockets | No | How many top-ranked P2Rank pockets to dock against. 1–20, default 3. |
| Vina exhaustiveness | No | Vina's search depth. 1–64, default 8. |
Supported file formats¶
Protein formats¶
| Extension | Description | Max size |
|---|---|---|
.pdb |
Protein Data Bank format | 50 MB |
.pdb.gz |
Gzipped PDB | 50 MB |
.cif |
mmCIF (macromolecular CIF) | 50 MB |
If your file fails the validation, the form returns:
Protein file must be PDB (.pdb), gzipped PDB (.pdb.gz), or mmCIF (.cif).
Ligand formats¶
| Extension | Description | Max size |
|---|---|---|
.sdf |
Structure-Data File | 10 MB |
.mol2 |
Tripos MOL2 | 10 MB |
.mol |
MDL Molfile | 10 MB |
PocketDock parses ligands with RDKit and prepares them for docking with Meeko. If your ligand can't be parsed, the job will fail with RDKit failed to read ligand file or Meeko failed to prepare the ligand molecule. See Troubleshooting for common causes.
Advanced settings¶
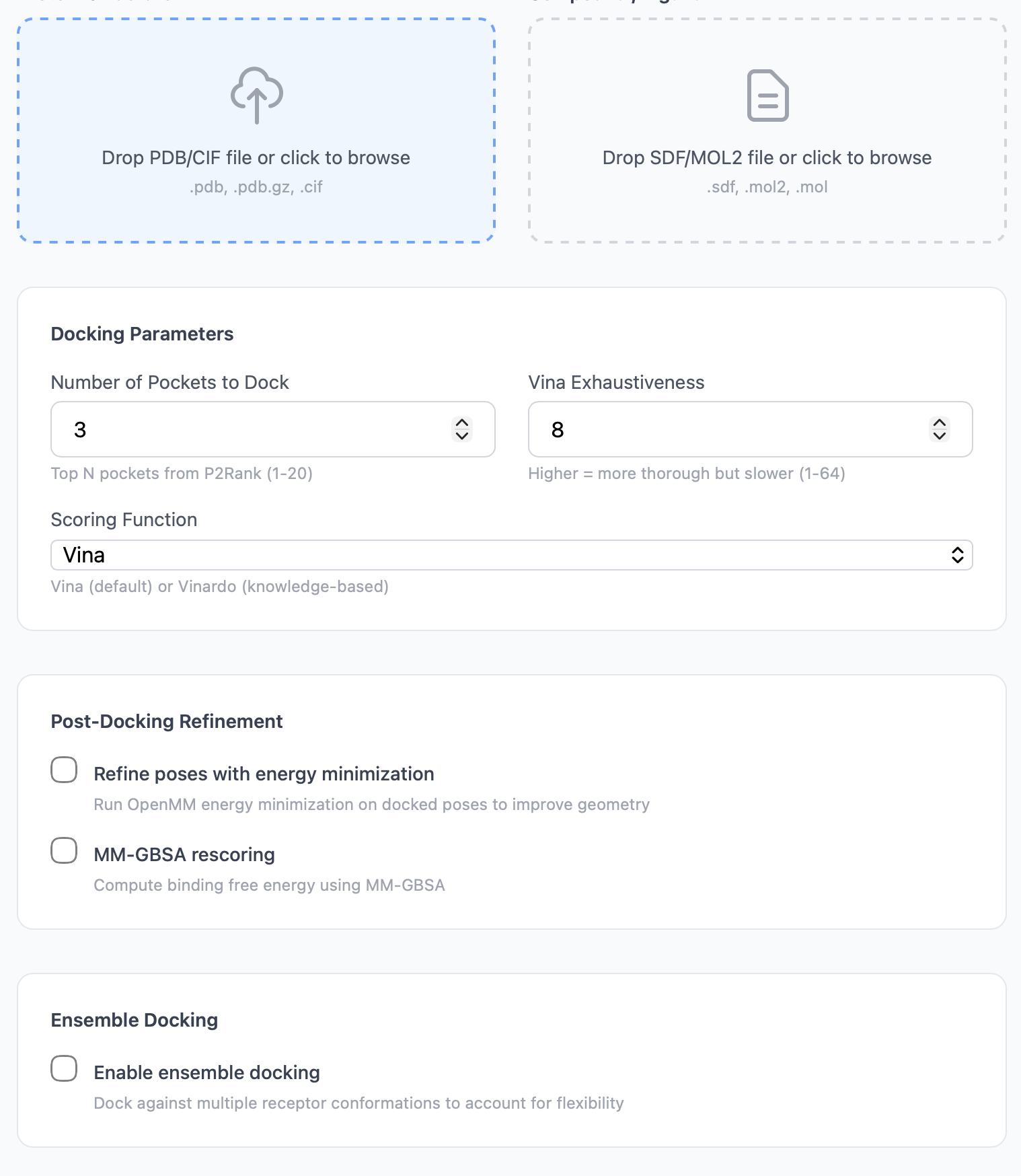
Number of pockets¶
Controls how many of P2Rank's predicted pockets to dock against. The pockets are ranked by P2Rank probability, so Number of pockets = 3 means "dock into the three most druggable predicted sites."
- Lower (1–2): Faster — useful when you already know roughly where the binding site is and just want a sanity check.
- Default (3): Reasonable trade-off between coverage and runtime.
- Higher (5–10+): Useful when the binding site is unknown or when you want to see how a ligand fares across multiple potential sites.
Each additional pocket adds one Vina run — total runtime scales roughly linearly with this parameter.
Vina exhaustiveness¶
Vina's exhaustiveness controls how many independent Monte Carlo + local-optimization runs are performed inside the docking box. Higher values reduce the chance of missing a low-energy pose at the cost of runtime.
- Default (8): Recommended for most jobs and screening workflows.
- 16: Use when the ligand is highly flexible (≥ 8 rotatable bonds) or when reproducibility matters across reruns.
- 32+: Use only when chasing the global minimum on a hard system. Runtime grows roughly linearly with exhaustiveness.
Drag-and-drop UX¶
Both file drop zones accept either drag-and-drop or click-to-browse. Once a file is dropped, its filename appears in the zone and the icon changes to indicate selection. To replace a file, drop a new one on top — the previous selection is replaced.
After submission¶
When you click Run Docking:
- The form is submitted as a multipart POST to
/api/jobs/. - The server creates a
DockingJobrecord, saves the files undermedia/jobs/<random-id>/, and queues a Celery task. - You're redirected to
/jobs/<job_id>/— the status page.
If the upload fails validation (wrong format, too large), you stay on the upload page with an error banner.
Tips¶
- Pre-clean your protein if it contains things you don't want docked against — alternate conformations, crystal waters, ions, ligands from the original crystal structure. PocketDock dockers against whatever atoms are in the file.
- Use a single chain when the biological assembly is a homodimer. Otherwise P2Rank may report symmetric pockets twice.
- Protonate the ligand at physiological pH before uploading. Vina assumes the protonation state in your file is correct.
- Center coordinates aren't needed — PocketDock derives the docking box automatically from each predicted pocket's geometry (controlled by the
VINA_BOX_PADDINGandVINA_DEFAULT_BOX_SIZEenv vars; see Configuration).