Ensemble Docking¶
Ensemble docking accounts for receptor flexibility by generating several plausible conformations of the protein and docking the ligand into all of them. PocketDock offers two flavors — a fast NMA mode and a thorough MD mode — and ranks results across the ensemble with a consensus score.
When to use it¶
- Flexible receptors — kinases, GPCRs, allosteric pockets, or any protein with mobile loops near the binding site.
- Induced-fit hypotheses — when the apo crystal structure may not represent the bound conformation.
- Hit triage — a ligand that docks well across multiple conformations is a more credible hit than one that only fits the rigid starting structure.
Single-conformation docking is fine when the receptor is rigid, the binding mode is well-characterized, or you're doing first-pass screening and runtime matters.
Enabling ensemble docking¶
On the upload page (single or batch tab), tick Enable ensemble docking and pick a method:
| Field | Notes |
|---|---|
| Ensemble method | NMA (default when enabled) or MD |
| Number of conformations | 2–10, default 5 |
The rest of the form (protein, ligand, pockets, exhaustiveness, optional refinement / MM-GBSA) is unchanged — those settings apply to every conformation in the ensemble.
NMA mode — fast and conservative¶
Method: Anisotropic Network Model (ANM) on the Cα atoms, implemented with NumPy/SciPy (no ProDy dependency).
Details:
- Hessian built from the Tirion harmonic potential with a 15 Å Cα–Cα cutoff and force constant γ = 1.0.
- Diagonalize and keep the 20 lowest non-trivial normal modes (or fewer for very small proteins).
- For each requested conformation, perturb the structure along a chosen mode with a scaled amplitude (1.5–3.0 Å Cα RMSD range), alternating sign.
- Output: N PDB files representing low-frequency, collective backbone motions of the receptor.
Runtime: roughly 30 seconds for 5 conformations on a typical kinase-sized protein. Scales linearly with num_conformations and atom count.
Use NMA when:
- You want a quick sense of how robust a docking result is to receptor breathing.
- The protein is large and MD-based sampling would be too slow.
- You're screening a batch and runtime per job matters.
NMA captures slow, collective motions but not local side-chain rearrangements — those need MD or explicit flexible-side-chain docking (not currently supported).
MD mode — thorough and slower¶
Method: short Langevin molecular dynamics with OpenMM and PDBFixer.
Details:
- PDBFixer cleans the input structure (adds missing residues and hydrogens).
- Force field: AMBER14-all + OBC2 implicit solvent.
- Energy minimization (max 200 iterations) before sampling.
- Langevin integrator at 300 K, 1 ps⁻¹ friction, 4 fs timestep with
HBondsconstraints. - Total simulation: 20 ps (5000 steps). N evenly-spaced snapshots are written as PDB files.
Runtime: roughly 5–15 minutes for 5 conformations, depending on protein size and CPU (no GPU acceleration in the default Docker image — see the optional celery-gpu service in docker-compose.yml).
Use MD when:
- The receptor has flexible loops that NMA can't capture (e.g., DFG flips in kinases).
- You suspect side-chain rearrangements in the pocket matter.
- You can afford the wall-clock time and want the most physically motivated conformations PocketDock can produce.
MD ensembles are short
20 ps is enough to sample local relaxation, not large-scale rearrangements. Treat the ensemble as "small perturbations around the starting structure," not as an exhaustive exploration of conformational space.
The ensemble pipeline¶
parent job (conformation_index = 0)
└─ generate N conformations (NMA or MD, status: running_ensemble)
├─ child 1 (conformation_index = 1) → P2Rank → Vina → results
├─ child 2 (conformation_index = 2) → P2Rank → Vina → results
├─ ...
└─ child N (conformation_index = N) → P2Rank → Vina → results
- The parent job acts as the coordinator. It generates the conformations, spawns children, and immediately transitions to
completed. - Each child is a normal
DockingJobwith its own protein PDB (the perturbed conformation), its own pockets, and its own docking results. - All jobs share an
ensemble_id(a 12-char UUID slice). - If you enable MM-GBSA rescoring or pose refinement, every child inherits those flags.
The ensemble dashboard¶
After submission you're redirected to /ensemble/<ensemble_id>/. The dashboard shows:
| Panel | Contents |
|---|---|
| Header | Ensemble name, protein and ligand filenames, method (NMA / MD), conformation count |
| Progress bar | Total / completed / failed / running / pending counts and a percentage |
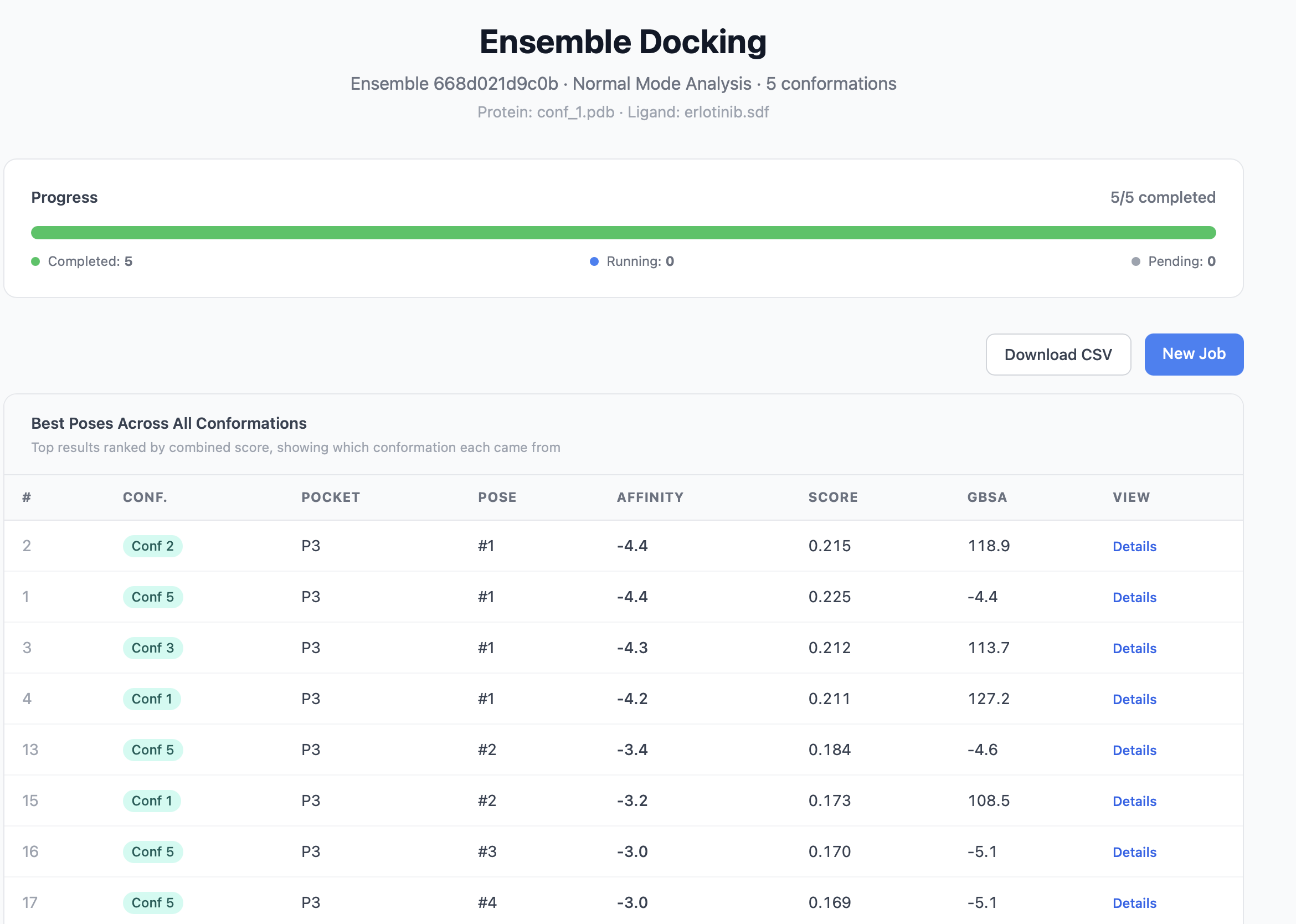
| Consensus top 20 | The 20 highest-scoring poses across all conformations, sorted by combined score. Each row shows which conformation it came from. |
| Per-conformation table | One row per child job: status, best affinity, best combined score, plus a top-10 pose list. |
The page auto-refreshes via /api/ensemble/<ensemble_id>/ (see the API reference).
Consensus scoring¶
PocketDock's consensus is straightforward: pool every pose from every conformation, sort by combined_score descending, and show the top 20.
This means:
- A pose that scores well in one conformation can win — useful for catching a binding mode that's only accessible to a specific receptor state.
- It does not penalize a pose for being missing in other conformations. If you want a "binds across the ensemble" metric, count how many of the top-N entries belong to each conformation, or fetch the full per-conformation top-10 from the dashboard.
Tips¶
- Start with NMA, especially in batch screening — get the runtime sense first.
- 5 conformations is a good default — enough to see whether docking is conformation-sensitive without quadrupling runtime.
- Eyeball the conformations — open the per-conformation pose page; if all five look identical, NMA's amplitude was too small for your system and MD is more useful.
- Combine with MM-GBSA when triaging a small hit list — but expect long runtimes (MD × N conformations × MM-GBSA rescoring).
- Don't ensemble-dock everything — if a single rigid run gives a clear, plausible pose with strong interactions, the ensemble usually confirms it without changing the answer.
See also¶
- Concepts: ensemble docking — terminology refresher
- API reference — ensemble endpoint — scripting ensemble runs
- Configuration — env vars that affect the per-conformation pipeline